
Ola Engkvist(オラ エンキビスト)氏
英AstraZeneca(AZ)社R&DのDiscovery Sciencesにおける分子AI部門の責任者。2つのバイオテクノロジー企業を経て、2004年にAZ社に入社。主な研究テーマは、ディープラーニングに基づく分子de novoデザイン、合成経路予測、大規模な分子特性予測。100以上の査読付き科学論文を発表している。スウェーデンのChalmers University of Technology(チャルマース工科大学)にてドラッグデザインにおける機械学習とAIの非常勤教授、英Cambridge Crystallographic Data Center(ケンブリッジ結晶学データセンター)の評議員も務めている。スウェーデンのLund University(ルンド大学)で計算化学の博士号を取得後、英University of Cambridgeでポスドクを務めた(記事内の画像はアストラゼネカが提供)
膨大な時間と費用を要する新薬の開発を人工知能(AI)で加速させるための研究が製薬業界で進んでいます。AI開発で必要とされるハードウエアとソフトウエアをプラットフォームとして提供する米NVIDIA社は現在、世界中の製薬企業と協業しており、その中で、英AstraZeneca(AZ)社とはAIモデルの開発においてコラボレーションを進めています。また、英国最大のスーパーコンピューターとして2021年に運用が開始されたCambridge-1の設立パートナーにもなっています。
2022年3月に開催されたNVIDIA社のグローバルカンファレンス「GTC 2022」では、スウェーデンにあるAZ社のヨーテボリ研究拠点でMolecular AI部門を率いるOla Engkvist(オラ エンキビスト)氏がセッションを行いました。「Accelerating Drug Design with AI(AIでドラッグデザインを加速させる)」と題した本セッションでは、AZ社における言語モデルを活用した最新の研究内容や、ドラッグデザインにAIを活用する上での成功とはどのようなことなのかを明かしており、本記事では、このセッションの内容の一部をご紹介します。
AIを活用するドラッグ デザインとは
エンキビスト氏は、AIを活用するドラッグ デザインとは「次にどの化合物を作るか」「どのように化合物を作るか」という2つの問いに対して効果的な答えを導きだそうとするものだと言います。一見、簡単そうに思えます。ですが最適な化合物を選択し、作ることは実際にはかなり難しいことなのです。
創薬において化合物の最適化プロセスは次のように始めます。まず出発点があります。ハイスループット スクリーニング、DNA-encoded libraryに基づくスクリーニング、フラグメントベースのスクリーニング、またはバーチャル スクリーニングなど化合物スクリーニングで確認された活性化合物を「ヒット化合物」と呼び、その後、「DMTA(Design・Make・Test・Analyse)」サイクルのステップを通して、化合物の最適化を行います。このサイクルを何度も何度も繰り返して、化合物を改良するのです。そして最終的に、動物モデルで高い効果を発揮し、代謝的に安定で、毒性に問題がないと考えられるドラッグの臨床候補を手に入れます。これには平均して少なくとも3年、あるいはそれ以上かかることもあります。
これに対して機械学習やAIを応用することでスピードアップを図ることが我々の野望です。具体的なゴールは、DMTAの1サイクルごとに得られる情報量を増やして全体のサイクル数を減らし、最も効果的な合成ルートを選択し、自動化技術の適用により1サイクル当たりの時間を短縮することです。
5年前はSF、でも今はそうではない
このような考え方は、5年前はサイエンスフィクション(SF)でした。しかしながらエンキビスト氏は「5年前と今とでは多くの違いがある」と語りました。NVIDIA社のGPUも、10年前に比べると演算能力が格段に上がり、新しいアーキテクチャーも登場しています。ニューラルネットワークのアルゴリズムも進歩しています。他分野の知見も取り入れられています。
さらに、画像認識、自然言語処理、オープンソース ソフトウエア等がイノベーションをさらに現実的なものにしています。
自然言語処理技術をドラッグデザインに応用する
画像のクリックで拡大表示
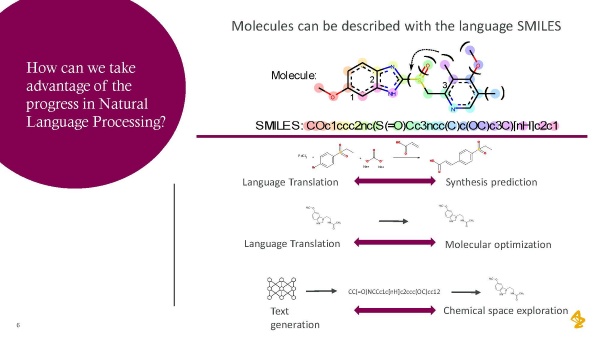
ケモインフォマティクス(Chemoinformatics)と自然言語処理の間には類似点があります。自然言語処理で開発された手法がドラッグデザインに活用できるのです。どうしてでしょうか。
分子の化学構造は文字列として、「SMILES(Simplified Molecular Input Line Entry System)」と呼ばれる直感的かつ曖昧性のない記述言語で表現することができます。SMILESには独自の文法があります。つまり自然言語処理のために開発されたツールが適用できるのです。分子の合成予測は、あたかも言語翻訳のように見ることができます。分子最適化も、出発分子から最終的な分子への翻訳として捉えることができます。つまり、自分が興味のある分子を関連するケミカルスペースに生成していくのです。
今できることは、以前とは何が違うのでしょうか? モンテカルロ木探索(注1、MCTS)などの新しいアルゴリズムによって、化合物の合成ルートを、より正確かつ高精度に予測できます。分子特性を予測するためにはグラフ畳み込みニューラルネットワークや様々なタイプのマルチタスク学習アルゴリズムなど、新しく、より柔軟な方法があります。蛋白質の構造を、AlphaFold2を通じてより高い精度で予測することも可能になっています。
注1)モンテカルロ木探索(MCTS)
木探索にモンテカルロ法(ランダム性)を使用するアルゴリズム。特に、囲碁、チェス、将棋などの対戦ゲームにおいて、次の有望な手を推定するために活用される。囲碁では、初めてプロ棋士に勝利したプログラム、AlphaGoでの探索に採用され貢献したことで知られる。
生成モデル vs. 列挙法
化学分野にディープラーニングを使うことは、とてもエキサイティングだとエンキビスト氏は言います。ディープラーニングのアーキテクチャを使うことで、関連するケミカルスペース全体をサンプリングすることができるからです。化合物の列挙によって化学空間をサンプリングしようとすると、10億から100億の化合物を格納するために何ギガバイト、何ペタバイトものメモリが必要になります。ですがディープラーニングを使ったアーキテクチャの場合、50メガバイト程度の容量で、化学物質空間全体をサンプリングすることができます。
画像のクリックで拡大表示
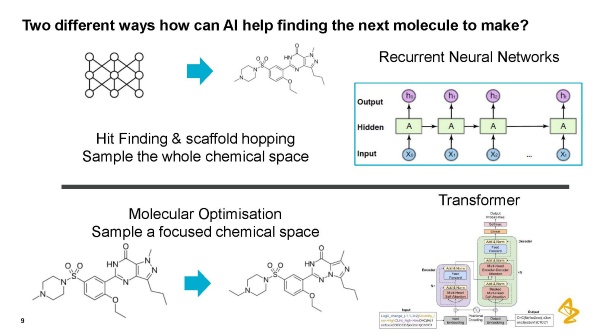
次に作るべき分子を見つけるためには2通りの方法があるとエンキビスト氏は言います。ポピュラーな方法の1つが再帰型ニューラルネットワーク(注2、以下RNN)を使うことです。他のニューラル ネットワークと同じように入力層と隠れ層、出力層がありますが、違うところは隠れノードにループが含まれており、過去の情報を利用して現在および将来の入力に対するネットワークの性能を向上させることができます。そのため、RNNは、スキャフォールド・ホッピングと呼ばれる、分子の大きな改変を行うのに適しています。
ですがエンキビスト氏は小さな変更を行って化合物を最適化したいのであれば別のアーキテクチャーを採用した方がよいかもしれない、と考えます。非常に効果的だったアーキテクチャーはTransformerでした。分子最適化には様々な方法があり、転移学習などを用いることもできますが、エンキビスト氏らが採った方法の中ではTransformerが一番適していたとのことです。
注2)再帰型ニューラルネットワーク(Recurrent Neural Network)
機械学習におけるニューラルネットワークの一種であり、一部の層の出力が、前の層の入力へと戻され、フィードバック ループが形成される。連続データの前後依存関係をうまく取り扱うことができるため、時系列データなど、前の情報がその次の情報に影響する連続的なデータを用いて機械学習を行う際に用いられる。
自然言語生成と化学分子合成の類似性
自然言語生成においてはテキストコーパスで学習した結果を利用して、文章で次に出てくる単語が何であるかを予測していきます。例えば「grasses(草)」という単語が出てくると、次にgreen(緑)かbrown(茶)という単語が出てくる確率が高いですが、purple(紫)という単語が出てくる確率は低い、といった具合です。この手法は基本的に、条件付き確率というコンセプトに基づいています。条件付き確率は、文中の単語に適用し、次の単語を予測することができるのです。
では、化学ではどうなるでしょうか。例えば最初に炭素があり、次の原子として炭素があって、さらに次に二重結合があるとすると、次の原子は何になるのでしょうか。また、その確率はどうなるのでしょうか。次の原子は、よく知られた化学物質であるホルムアルデヒドが生成される酸素である確率が最も高くなります。このようにケミカルスペースでのサンプリングにおいて条件付き確率を使うための手掛かりを教えてくれるのです。
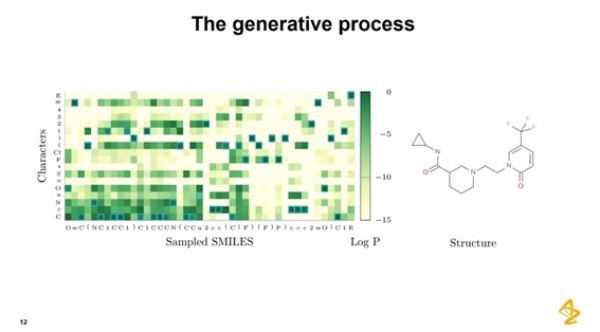
エンキビスト氏は、学習セットには存在しないけれども、原理的には学習セットに存在し得る化合物を生成する例を示しました。SMILESの文字列の各文字が、ある確率で選択されます。炭素や酸素などの原子(文字)の確率が表示されるので、目的に合わせて選択していくと、分子構造が表示されるというものです。条件付き確率の分布の様子が色分けされて明示されます。このように分子を積み上げていくことで、コンピューターの中で分子を効率的にサンプリングしていくことができるのです。
機械学習活用は「百聞は一見にしかず」
エンキビスト氏らが開発した「REINVENT 2.0」のようなディープラーニングを使った分子生成ツールはオープンソース化されており、誰でも利用できます。エンキビスト氏は「百聞は一見にしかず。ぜひご自身でソフトウエアツールを試してみてください」と語りました。「ドラッグデザインにおける機械学習の活用については様々な意見がありますが、重要なことは実際に自分で試してみて、どんな分子が生成されるのかを確認することです」
エンキビスト氏らはAiZynthFinderという逆合成解析ツール、合成ルート予測ツールも開発しています。世界中でそれを実際に試している人たちがいます。例えば「逆合成ちゃん(@retrosynthchan)」というTwitterボットは、SMILESをリプライするとAiZynthFinderによる逆合成(合成経路を探すための手法)の結果を返してくれます。
成功とはどのようなものか?
最後にエンキビスト氏は、機械学習を用いた創薬の成功とは、どのようなものかを示しました。機械学習を使うことで創薬の時間を短縮することはできます。ですが、エンキビスト氏によると、この指標は「成功」そのものではなく、「成功の結果」に近い。では何が成功なのでしょうか。機械学習やAIを適用する他の分野と同じで「信頼を得ること」だと言います。
創薬分野においても、AIが設計した分子が、X線結晶構造解析と同等の信頼を得ること、それが成功だというのです。創薬における機械学習の信頼には、2つあります。1つは、個々の分子に対する予測への信頼。もう1つは、機械学習/AIによって生成された分子が、プロジェクト全体の進行を加速させることができるという信頼です。AIが生成した分子が、最も効率的に臨床候補に導ける最適な分子である、という信頼を得ること。それが成功だとエンキビスト氏は語りました。
著者紹介
平畠浩司
エヌビディア合同会社 エンタープライズ事業部 ライフサイエンス事業開発担当
2005年より国内大手製薬会社にてセールスおよびマーケティングに従事。その後、海外バイオテクノロジー企業の開発パイプラインの導入・導出の事業開発アドバイザー、医療AIベンチャー企業を経て、2021年にNVIDIAに入社。NVIDIAでは創薬領域をはじめとしたライフサイエンスにおけるAI活用の活性化を推進
山田泰永
エヌビディア合同会社 エンタープライズ事業部 メディカル・ライフサイエンス領域マネージャー兼 AIスタートアップ・技術パートナー支援担当
1998年より日系半導体メーカーや商社にてセールスおよびプロダクトマーケティングに従事。2004年にNVIDIAに入社し、PC/ワークステーション向けGPUやタブレット端末向けTegra SoCのセールスやプロジェクトサポートを担当。2015年より組み込み用途向けのディープラーニング、さらに2016年よりメディカル・ライフサイエンス領域のビジネス開拓を担当。また、AIスタートアップ企業の支援や連携によるエコシステム構築も推進
からの記事と詳細 ( ドラッグデザインにおけるAI活用、少し前ならSFだった - 日経バイオテク )
https://ift.tt/jXWTc2J
No comments:
Post a Comment